Acetylation-dependent interaction of SATB1 and CtBP1 mediates transcriptional repression by SATB1.
Purbey, Prabhat Kumar, et al.
Mol. Cell. Biol., 29: 1321-37 (2009)
2009
| 19103759
 |
Splicing of HDAC7 modulates the SRF-myocardin complex during stem-cell differentiation towards smooth muscle cells.
Margariti, Andriana, et al.
J. Cell. Sci., 122: 460-70 (2009)
2009
Show Abstract
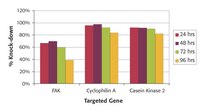
Histone deacetylases (HDACs) have a central role in the regulation of gene expression. Here we investigated whether HDAC7 has an impact on embryonic stem (ES) cell differentiation into smooth muscle cells (SMCs). ES cells were seeded on collagen-IV-coated flasks and cultured in the absence of leukemia inhibitory factor in differentiation medium to induce SMC differentiation. Western blots and double-immunofluorescence staining demonstrated that HDAC7 has a parallel expression pattern with SMC marker genes. In ex vivo culture of embryonic cells from SM22-LacZ transgenic mice, overexpression of HDAC7 significantly increased beta-galactosidase-positive cell numbers and enzyme activity, indicating its crucial role in SMC differentiation during embryonic development. We found that HDAC7 undergoes alternative splicing during ES cell differentiation. Platelet-derived growth factor enhanced ES cell differentiation into SMCs through upregulation of HDAC7 splicing. Further experiments revealed that HDAC7 splicing induced SMC differentiation through modulation of the SRF-myocardin complex. These findings suggest that HDAC7 splicing is important for SMC differentiation and vessel formation in embryonic development. | 19174469
|
CREB mediates prostaglandin F2alpha-induced MUC5AC overexpression.
Chung, Wen-Cheng, et al.
J. Immunol., 182: 2349-56 (2009)
2009
| 19201889
|
Tumor necrosis factor alpha regulates responses to nerve growth factor, promoting neural cell survival but suppressing differentiation of neuroblastoma cells.
Takei, Yoshinori and Laskey, Ronald
Mol. Biol. Cell, 19: 855-64 (2008)
2008
| 18094051
|
Transcription factor FOXO3a mediates apoptosis in HIV-1-infected macrophages.
Cui, Min, et al.
J. Immunol., 180: 898-906 (2008)
2008
Show Abstract
Macrophages serve as a major reservoir for HIV-1 because a large number of macrophages in the brain and lung are infected with HIV-1 during late stage disease. Recent evidence suggests that those HIV-1-infected macrophages play a key role in contributing to tissue damage in AIDS pathogenesis. Macrophages undergo apoptosis upon HIV-1 infection; however, the mechanisms of this process are not well-defined. Previously, we demonstrated that HIV-1 infection inhibits Akt-1, a critical protein for cell survival of macrophages. In the present study, we investigated the involvement of transcription factor FOXO3a in the regulation of HIV-1-mediated apoptosis in macrophages. HIV-1 infection significantly decreased phosphorylation of FOXO3a and promoted FOXO3a translocation to the nucleus in human monocyte-derived macrophages (MDM). Overexpression of a constitutively active FOXO3a increased DNA fragmentation with decreased cell viability in MDM, whereas a dominant-negative mutant of FOXO3a or small interfering RNA for FOXO3a to knockdown the function of FOXO3a in HIV-1-infected MDM decreased DNA fragmentation and protected macrophages from death in HIV-1-infected MDM. Overexpression of constitutively active Akt-1 increased FOXO3a phosphorylation, suggesting that FOXO3a phosphorylation in human MDM is dependent on Akt-1. We therefore conclude that FOXO3a plays an important role in HIV-1-induced cell death of human macrophage. Understanding the PI3K/Akt-1/FOXO3a pathway and its associated death mechanism in macrophages during HIV-1 infection would lead to identification of potential therapeutic avenues for the treatment of HIV-1 infection. | 18178829
|
Growth suppression of lung cancer cells by targeting cyclic AMP response element-binding protein.
Aggarwal, Sita, et al.
Cancer Res., 68: 981-8 (2008)
2008
Show Abstract
Genes regulated by cyclic AMP-response element-binding protein (CREB) have been reported to suppress apoptosis, induce cell proliferation, and mediate inflammation and tumor metastasis. However, it is not clear whether CREB is critically involved in lung carcinogenesis. We found that non-small cell lung cancer (NSCLC) cell lines exhibited elevated constitutive activity in CREB, in its immediate upstream kinases (ribosomal s6 kinase and extracellular signal kinase), and in the CREB-regulated cell survival proteins Bcl-2 and Bcl-xL. We hypothesized that constitutively active CREB is important to lung cancer cell growth and survival and therefore could be a potential therapeutic target for NSCLC. Ectopic expression of dominant repressor CREB and transfection with small interfering RNA against CREB suppressed the growth and survival of NSCLC cells and induced apoptotic cell death. Furthermore, treating H1734 NSCLC cells with an inhibitor of the CREB signaling pathway Ro-31-8220 inhibited CREB activation by blocking the activity of extracellular signal kinase and ribosomal s6 kinase, arrested the cell cycle at the G(2)-M phase, and subsequently induced apoptosis with the suppression of Bcl-2 and Bcl-xL expression. Ro-31-8220 suppressed both the anchorage-dependent and independent growth of NSCLC cells, but its cytotoxic effect was much less prominent in normal bronchial epithelial cells. Our results indicate that active CREB plays an important role in NSCLC cell growth and survival. Thus, agents that suppress CREB activation could have potential therapeutic value for NSCLC treatment. | 18281471
|
Protein phosphatase 2A negatively regulates integrin alpha(IIb)beta(3) signaling.
Gushiken, Francisca C, et al.
J. Biol. Chem., 283: 12862-9 (2008)
2008
| 18334487
|
Prolactin and ErbB4/HER4 signaling interact via Janus kinase 2 to induce mammary epithelial cell gene expression differentiation.
Muraoka-Cook, Rebecca S, et al.
Mol. Endocrinol., 22: 2307-21 (2008)
2008
Show Abstract
Differentiation of mammary epithelium in vivo requires signaling through prolactin and ErbB4/HER4-dependent mechanisms. Although stimulation of either the prolactin receptor or ErbB4/HER4 results in activation of the transcription factor signal transducer and activator of transcription 5A (STAT5A) and induction of lactogenic differentiation, how these pathways intersect is unknown. We show herein that prolactin signaling in breast cells cooperates with and is substantially enhanced by the receptor tyrosine kinase ErbB4/HER4. Prolactin and the ErbB4/HER4 ligand heparin-binding epidermal growth factor each induced STAT5A tyrosine phosphorylation and nuclear translocation; each pathway required the intracellular tyrosine kinase Janus kinase 2 (JAK2). We found that full prolactin-mediated STAT5A activation and binding to the endogenous beta-casein promoter required ErbB4/HER4 but did not require ErbB1/epidermal growth factor receptor. For example, prolactin-induced STAT5A activity was markedly diminished in cells overexpressing kinase inactive HER4, in cells transfected with small interfering RNAs to specifically knock down endogenous ErbB4/HER4 expression and in cells treated with a small molecule inhibitor that targets ErbB4 kinase. Interestingly, prolactin caused ErbB4/HER4 tyrosine phosphorylation in a JAK2 kinase-dependent manner. Finally, prolactin receptor, ErbB4/HER4, and JAK2 were coimmunoprecipitated from prolactin-treated but not untreated cells. These results suggest that prolactin signaling engages the ErbB4 pathway via JAK2 and that ErbB4 provides an important component of STAT5A-dependent lactogenic differentiation; this pathway integration may help explain the similar deficit in mammary development observed in gene-targeted mice deficient in prolactin receptor, JAK2, ErbB4, or STAT5A. | 18653779
|
The HER4/4ICD estrogen receptor coactivator and BH3-only protein is an effector of tamoxifen-induced apoptosis.
Naresh, Anjali, et al.
Cancer Res., 68: 6387-95 (2008)
2008
| 18676864
|
CD40 induces antigen transporter and immunoproteasome gene expression in carcinomas via the coordinated action of NF-kappaB and of NF-kappaB-mediated de novo synthesis of IRF-1.
Moschonas, Aristides, et al.
Mol. Cell. Biol., 28: 6208-22 (2008)
2008
| 18694960
|