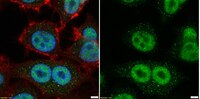
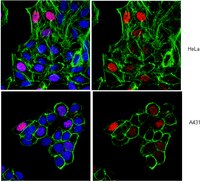
Analysis of Histones H3 and H4 Reveals Novel and Conserved Post-Translational Modifications in Sugarcane.
Moraes, I; Yuan, ZF; Liu, S; Souza, GM; Garcia, BA; Casas-Mollano, JA
PloS one
10
e0134586
2015
Show Abstract
Histones are the main structural components of the nucleosome, hence targets of many regulatory proteins that mediate processes involving changes in chromatin. The functional outcome of many pathways is "written" in the histones in the form of post-translational modifications that determine the final gene expression readout. As a result, modifications, alone or in combination, are important determinants of chromatin states. Histone modifications are accomplished by the addition of different chemical groups such as methyl, acetyl and phosphate. Thus, identifying and characterizing these modifications and the proteins related to them is the initial step to understanding the mechanisms of gene regulation and in the future may even provide tools for breeding programs. Several studies over the past years have contributed to increase our knowledge of epigenetic gene regulation in model organisms like Arabidopsis, yet this field remains relatively unexplored in crops. In this study we identified and initially characterized histones H3 and H4 in the monocot crop sugarcane. We discovered a number of histone genes by searching the sugarcane ESTs database. The proteins encoded correspond to canonical histones, and their variants. We also purified bulk histones and used them to map post-translational modifications in the histones H3 and H4 using mass spectrometry. Several modifications conserved in other plants, and also novel modified residues, were identified. In particular, we report O-acetylation of serine, threonine and tyrosine, a recently identified modification conserved in several eukaryotes. Additionally, the sub-nuclear localization of some well-studied modifications (i.e., H3K4me3, H3K9me2, H3K27me3, H3K9ac, H3T3ph) is described and compared to other plant species. To our knowledge, this is the first report of histones H3 and H4 as well as their post-translational modifications in sugarcane, and will provide a starting point for the study of chromatin regulation in this crop. | | | 26226299
 |
Progenitor stage-specific activity of a cis-acting double GATA motif for Gata1 gene expression.
Moriguchi, T; Suzuki, M; Yu, L; Takai, J; Ohneda, K; Yamamoto, M
Molecular and cellular biology
35
805-15
2015
Show Abstract
GATA1 is a master regulator of erythropoiesis, expression of which is regulated by multiple discrete cis-acting elements. In this study, we examine the activity of a promoter-proximal double GATA (dbGATA) motif, using a Gata1 bacterial artificial chromosome (BAC)-transgenic green fluorescent protein (GFP) reporter (G1BAC-GFP) mouse system. Deletion of the dbGATA motif led to significant reductions in GFP expression in hematopoietic progenitors, while GFP expression was maintained in erythroblasts. Consistently, in mice with a germ line deletion of the dbGATA motif (Gata1(ΔdbGATA) mice), GATA1 expression in progenitors was significantly decreased. The suppressed GATA1 expression was associated with a compensatory increase in GATA2 levels in progenitors. When we crossed Gata1(ΔdbGATA) mice with Gata2 hypomorphic mutant mice (Gata2(fGN/fGN) mice), the Gata1(ΔdbGATA)::Gata2(fGN/fGN) compound mutant mice succumbed to a significant decrease in the progenitor population, whereas both groups of single mutant mice maintained progenitors and survived to adulthood, indicating the functional redundancy between GATA1 and GATA2 in progenitors. Meanwhile, the effects of the dbGATA site deletion on Gata1 expression were subtle in erythroblasts, which showed increased GATA1 binding and enhanced accumulation of active histone marks around the 1st-intron GATA motif of the ΔdbGATA locus. These results thus reveal a novel role of the dbGATA motif in the maintenance of Gata1 expression in hematopoietic progenitors and a functional compensation between the dbGATA site and the 1st-intron GATA motif in erythroblasts. | | | 25535330
|
Immediate chromatin immunoprecipitation and on-bead quantitative PCR analysis: a versatile and rapid ChIP procedure.
Harmeyer, KM; South, PF; Bishop, B; Ogas, J; Briggs, SD
Nucleic acids research
43
e38
2015
Show Abstract
Genome-wide chromatin immunoprecipitation (ChIP) studies have brought significant insight into the genomic localization of chromatin-associated proteins and histone modifications. The large amount of data generated by these analyses, however, require approaches that enable rapid validation and analysis of biological relevance. Furthermore, there are still protein and modification targets that are difficult to detect using standard ChIP methods. To address these issues, we developed an immediate chromatin immunoprecipitation procedure which we call ZipChip. ZipChip significantly reduces the time and increases sensitivity allowing for rapid screening of multiple loci. Here we describe how ZipChIP enables detection of histone modifications (H3K4 mono- and trimethylation) and two yeast histone demethylases, Jhd2 and Rph1, which were previously difficult to detect using standard methods. Furthermore, we demonstrate the versatility of ZipChIP by analyzing the enrichment of the histone deacetylase Sir2 at heterochromatin in yeast and enrichment of the chromatin remodeler, PICKLE, at euchromatin in Arabidopsis thaliana. | | | 25539918
|
Incorporation of histone H3.1 suppresses the lineage potential of skeletal muscle.
Harada, A; Maehara, K; Sato, Y; Konno, D; Tachibana, T; Kimura, H; Ohkawa, Y
Nucleic acids research
43
775-86
2015
Show Abstract
Lineage potential is triggered by lineage-specific transcription factors in association with changes in the chromatin structure. Histone H3.3 variant is thought to play an important role in the regulation of lineage-specific genes. To elucidate the function of H3.3 in myogenic differentiation, we forced the expression of GFP-H3.1 to alter the balance between H3.1 and H3.3 in mouse C2C12 cells that could be differentiated into myotubes. GFP-H3.1 replaced H3.3 in the regulatory regions of skeletal muscle (SKM) genes and induced a decrease of H3K4 trimethylation (H3K4me3) and increase of H3K27 trimethylation (H3K27me3). Similar results were obtained by H3.3 knockdown. In contrast, MyoD-dependent H3.3 incorporation into SKM genes in fibroblasts induced an increase of H3K4me3 and H3K27me3. In mouse embryos, a bivalent modification of H3K4me3 and H3K27me3 was formed on H3.3-incorporated SKM genes before embryonic skeletal muscle differentiation. These results suggest that lineage potential is established through a selective incorporation of specific H3 variants that governs the balance of histone modifications. | | | 25539924
|
The oncometabolite D-2-hydroxyglutarate induced by mutant IDH1 or -2 blocks osteoblast differentiation in vitro and in vivo.
Suijker, J; Baelde, HJ; Roelofs, H; Cleton-Jansen, AM; Bovée, JV
Oncotarget
6
14832-42
2015
Show Abstract
Mutations in isocitrate dehydrogenase 1 (IDH1) and IDH2 are found in a somatic mosaic fashion in patients with multiple enchondromas. Enchondromas are benign cartilaginous tumors arising in the medulla of bone. The mutant IDH1/2 causes elevated levels of D-2-hydroxyglutarate (D-2-HG). Mesenchymal stem cells (MSC) are the precursor of the osteoblastic, chondrogenic and adipocytic lineage and we hypothesized that increased levels of D-2-HG cause multiple enchondromas by affecting differentiation of MSCs. Bone marrow derived MSCs from different donors were differentiated towards osteoblastic, chondrogenic and adipocytic lineage in the presence or absence of 5 mM D-2-HG. Three of four MSCs showed near complete inhibition of calcification after 3 weeks under osteogenic differentiation conditions in the presence of D-2-HG, indicating a block in osteogenic differentiation. Two of four MSCs showed an increase in differentiation towards the chondrogenic lineage. To evaluate the effect of D-2-HG in vivo we monitored bone development in zebrafish, which revealed an impaired development of vertebrate rings in the presence of D-2-HG compared to control conditions (p-value less than 0.0001). Our data indicate that increased levels of D-2-HG promote chondrogenic over osteogenic differentiation. Thus, mutations in IDH1/2 lead to a local block in osteogenic differentiation during skeletogenesis causing the development of benign cartilaginous tumors. | | | 26046462
|
Epigenetic Modifications of the PGC-1α Promoter during Exercise Induced Expression in Mice.
Lochmann, TL; Thomas, RR; Bennett, JP; Taylor, SM
PloS one
10
e0129647
2015
Show Abstract
The transcriptional coactivator, PGC-1α, is known for its role in mitochondrial biogenesis. Although originally thought to exist as a single protein isoform, recent studies have identified additional promoters which produce multiple mRNA transcripts. One of these promoters (promoter B), approximately 13.7 kb upstream of the canonical PGC-1α promoter (promoter A), yields alternative transcripts present at levels much lower than the canonical PGC-1α mRNA transcript. In skeletal muscle, exercise resulted in a substantial, rapid increase of mRNA of these alternative PGC-1α transcripts. Although the β2-adrenergic receptor was identified as a signaling pathway that activates transcription from PGC-1α promoter B, it is not yet known what molecular changes occur to facilitate PGC-1α promoter B activation following exercise. We sought to determine whether epigenetic modifications were involved in this exercise response in mouse skeletal muscle. We found that DNA hydroxymethylation correlated to increased basal mRNA levels from PGC-1α promoter A, but that DNA methylation appeared to play no role in the exercise-induced activation of PGC-1α promoter B. The level of the activating histone mark H3K4me3 increased with exercise 2-4 fold across PGC-1α promoter B, but remained unaltered past the canonical PGC-1α transcriptional start site. Together, these data show that epigenetic modifications partially explain exercise-induced changes in the skeletal muscle mRNA levels of PGC-1α isoforms. | | | 26053857
|
miR-142-5p and miR-130a-3p are regulated by IL-4 and IL-13 and control profibrogenic macrophage program.
Su, S; Zhao, Q; He, C; Huang, D; Liu, J; Chen, F; Chen, J; Liao, JY; Cui, X; Zeng, Y; Yao, H; Su, F; Liu, Q; Jiang, S; Song, E
Nature communications
6
8523
2015
Show Abstract
Macrophages play a pivotal role in tissue fibrogenesis, which underlies the pathogenesis of many end-stage chronic inflammatory diseases. MicroRNAs are key regulators of immune cell functions, but their roles in macrophage's fibrogenesis have not been characterized. Here we show that IL-4 and IL-13 induce miR-142-5p and downregulate miR-130a-3p in macrophages; these changes sustain the profibrogenic effect of macrophages. In vitro, miR-142-5p mimic prolongs STAT6 phosphorylation by targeting its negative regulator, SOCS1. Blocking miR-130a relieves its inhibition of PPARγ, which coordinates STAT6 signalling. In vivo, inhibiting miR-142-5p and increasing miR-130a-3p expression with locked nucleic acid-modified oligonucleotides inhibits CCL4-induced liver fibrosis and bleomycin-induced lung fibrosis in mice. Furthermore, macrophages from the tissue samples of patients with liver cirrhosis and idiopathic pulmonary fibrosis display increased miR-142-5p and decreased miR-130a-3p expression. Therefore, miR-142-5p and miR-130a-3p regulate macrophage profibrogenic gene expression in chronic inflammation. | | | 26436920
|
Genome-wide mapping of histone H3 lysine 4 trimethylation in Eucalyptus grandis developing xylem.
Hussey, SG; Mizrachi, E; Groover, A; Berger, DK; Myburg, AA
BMC plant biology
15
117
2015
Show Abstract
Histone modifications play an integral role in plant development, but have been poorly studied in woody plants. Investigating chromatin organization in wood-forming tissue and its role in regulating gene expression allows us to understand the mechanisms underlying cellular differentiation during xylogenesis (wood formation) and identify novel functional regions in plant genomes. However, woody tissue poses unique challenges for using high-throughput chromatin immunoprecipitation (ChIP) techniques for studying genome-wide histone modifications in vivo. We investigated the role of the modified histone H3K4me3 (trimethylated lysine 4 of histone H3) in gene expression during the early stages of wood formation using ChIP-seq in Eucalyptus grandis, a woody biomass model.Plant chromatin fixation and isolation protocols were optimized for developing xylem tissue collected from field-grown E. grandis trees. A "nano-ChIP-seq" procedure was employed for ChIP DNA amplification. Over 9 million H3K4me3 ChIP-seq and 18 million control paired-end reads were mapped to the E. grandis reference genome for peak-calling using Model-based Analysis of ChIP-Seq. The 12,177 significant H3K4me3 peaks identified covered ~1.5% of the genome and overlapped some 9,623 protein-coding genes and 38 noncoding RNAs. H3K4me3 library coverage, peaking ~600 - 700 bp downstream of the transcription start site, was highly correlated with gene expression levels measured with RNA-seq. Overall, H3K4me3-enriched genes tended to be less tissue-specific than unenriched genes and were overrepresented for general cellular metabolism and development gene ontology terms. Relative expression of H3K4me3-enriched genes in developing secondary xylem was higher than unenriched genes, however, and highly expressed secondary cell wall-related genes were enriched for H3K4me3 as validated using ChIP-qPCR.In this first genome-wide analysis of a modified histone in a woody tissue, we optimized a ChIP-seq procedure suitable for field-collected samples. In developing E. grandis xylem, H3K4me3 enrichment is an indicator of active transcription, consistent with its known role in sustaining pre-initiation complex formation in yeast. The H3K4me3 ChIP-seq data from this study paves the way to understanding the chromatin landscape and epigenomic architecture of xylogenesis in plants, and complements RNA-seq evidence of gene expression for the future improvement of the E. grandis genome annotation. | | | 25957781
|
Negative regulatory roles of DE-ETIOLATED1 in flowering time in Arabidopsis.
Kang, MY; Yoo, SC; Kwon, HY; Lee, BD; Cho, JN; Noh, YS; Paek, NC
Scientific reports
5
9728
2015
Show Abstract
Arabidopsis flowers early under long days (LD) and late under short days (SD). The repressor of photomorphogenesis DE-ETIOLATED1 (DET1) delays flowering; det1-1 mutants flower early, especially under SD, but the molecular mechanism of DET1 regulation remains unknown. Here we examine the regulatory function of DET1 in repression of flowering. Under SD, the det1-1 mutation causes daytime expression of FKF1 and CO; however, their altered expression has only a small effect on early flowering in det1-1 mutants. Notably, DET1 interacts with GI and binding of GI to the FT promoter increases in det1-1 mutants, suggesting that DET1 mainly restricts GI function, directly promoting FT expression independent of CO expression. Moreover, DET1 interacts with MSI4/FVE, which epigenetically inhibits FLC expression, indicating that the lack of FLC expression in det1-1 mutants likely involves altered histone modifications at the FLC locus. These data demonstrate that DET1 acts in both photoperiod and autonomous pathways to inhibit expression of FT and SOC1. Consistent with this, the early flowering of det1-1 mutants disappears completely in the ft-1 soc1-2 double mutant background. Thus, we propose that DET1 is a strong repressor of flowering and has a pivotal role in maintaining photoperiod sensitivity in the regulation of flowering time. | | | 25962685
|
Conserved Epigenetic Mechanisms Could Play a Key Role in Regulation of Photosynthesis and Development-Related Genes during Needle Development of Pinus radiata.
Valledor, L; Pascual, J; Meijón, M; Escandón, M; Cañal, MJ
PloS one
10
e0126405
2015
Show Abstract
Needle maturation is a complex process that involves cell growth, differentiation and tissue remodelling towards the acquisition of full physiological competence. Leaf induction mechanisms are well known; however, those underlying the acquisition of physiological competence are still poorly understood, especially in conifers. We studied the specific epigenetic regulation of genes defining organ function (PrRBCS and PrRBCA) and competence and stress response (PrCSDP2 and PrSHMT4) during three stages of needle development and one de-differentiated control. Gene-specific changes in DNA methylation and histone were analysed by bisulfite sequencing and chromatin immunoprecipitation (ChIP). The expression of PrRBCA and PrRBCS increased during needle maturation and was associated with the progressive loss of H3K9me3, H3K27me3 and the increase in AcH4. The maturation-related silencing of PrSHMT4 was correlated with increased H3K9me3 levels, and the repression of PrCSDP2, to the interplay between AcH4, H3K27me3, H3K9me3 and specific DNA methylation. The employ of HAT and HDAC inhibitors led to a further determination of the role of histone acetylation in the regulation of our target genes. The integration of these results with high-throughput analyses in Arabidopsis thaliana and Populus trichocarpa suggests that the specific epigenetic mechanisms that regulate photosynthetic genes are conserved between the analysed species. | | | 25965766
|