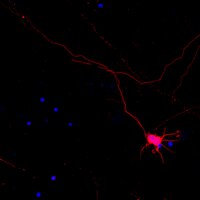
Synaptic dysregulation in a human iPS cell model of mental disorders.
Wen, Z; Nguyen, HN; Guo, Z; Lalli, MA; Wang, X; Su, Y; Kim, NS; Yoon, KJ; Shin, J; Zhang, C; Makri, G; Nauen, D; Yu, H; Guzman, E; Chiang, CH; Yoritomo, N; Kaibuchi, K; Zou, J; Christian, KM; Cheng, L; Ross, CA; Margolis, RL; Chen, G; Kosik, KS; Song, H; Ming, GL
Nature
2014
Show Abstract
Dysregulated neurodevelopment with altered structural and functional connectivity is believed to underlie many neuropsychiatric disorders, and 'a disease of synapses' is the major hypothesis for the biological basis of schizophrenia. Although this hypothesis has gained indirect support from human post-mortem brain analyses and genetic studies, little is known about the pathophysiology of synapses in patient neurons and how susceptibility genes for mental disorders could lead to synaptic deficits in humans. Genetics of most psychiatric disorders are extremely complex due to multiple susceptibility variants with low penetrance and variable phenotypes. Rare, multiply affected, large families in which a single genetic locus is probably responsible for conferring susceptibility have proven invaluable for the study of complex disorders. Here we generated induced pluripotent stem (iPS) cells from four members of a family in which a frameshift mutation of disrupted in schizophrenia 1 (DISC1) co-segregated with major psychiatric disorders and we further produced different isogenic iPS cell lines via gene editing. We showed that mutant DISC1 causes synaptic vesicle release deficits in iPS-cell-derived forebrain neurons. Mutant DISC1 depletes wild-type DISC1 protein and, furthermore, dysregulates expression of many genes related to synapses and psychiatric disorders in human forebrain neurons. Our study reveals that a psychiatric disorder relevant mutation causes synapse deficits and transcriptional dysregulation in human neurons and our findings provide new insight into the molecular and synaptic etiopathology of psychiatric disorders. | | 25132547
 |
Endogenous cerebellar neurogenesis in adult mice with progressive ataxia.
Kumar, M; Csaba, Z; Peineau, S; Srivastava, R; Rasika, S; Mani, S; Gressens, P; El Ghouzzi, V
Annals of clinical and translational neurology
1
968-81
2014
Show Abstract
Transplanting exogenous neuronal progenitors to replace damaged neurons in the adult brain following injury or neurodegenerative disorders and achieve functional amelioration is a realistic goal. However, studies so far have rarely taken into consideration the preexisting inflammation triggered by the disease process that could hamper the effectiveness of transplanted cells. Here, we examined the fate and long-term consequences of human cerebellar granule neuron precursors (GNP) transplanted into the cerebellum of Harlequin mice, an adult model of progressive cerebellar degeneration with early-onset microgliosis.Human embryonic stem cell-derived progenitors expressing Atoh1, a transcription factor key to GNP specification, were generated in vitro and stereotaxically transplanted into the cerebellum of preataxic Harlequin mice. The histological and functional impact of these transplants was followed using immunolabeling and Rotarod analysis.Although transplanted GNPs did not survive beyond a few weeks, they triggered the proliferation of endogenous nestin-positive precursors in the leptomeninges that crossed the molecular layer and differentiated into mature neurons. These phenomena were accompanied by the preservation of the granule and Purkinje cell layers and delayed ataxic changes. In vitro neurosphere generation confirmed the enhanced neurogenic potential of the cerebellar leptomeninges of Harlequin mice transplanted with exogenous GNPs.The cerebellar leptomeninges of adult mice contain an endogenous neurogenic niche that can be stimulated to yield mature neurons from an as-yet unidentified population of progenitors. The transplantation of human GNPs not only stimulates this neurogenesis, but, despite the potentially hostile environment, leads to neuroprotection and functional amelioration. | Immunofluorescence | 25574472
|
Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa.
Tucker, BA; Mullins, RF; Streb, LM; Anfinson, K; Eyestone, ME; Kaalberg, E; Riker, MJ; Drack, AV; Braun, TA; Stone, EM
eLife
2
e00824
2013
Show Abstract
Next-generation and Sanger sequencing were combined to identify disease-causing USH2A mutations in an adult patient with autosomal recessive RP. Induced pluripotent stem cells (iPSCs), generated from the patient's keratinocytes, were differentiated into multi-layer eyecup-like structures with features of human retinal precursor cells. The inner layer of the eyecups contained photoreceptor precursor cells that expressed photoreceptor markers and exhibited axonemes and basal bodies characteristic of outer segments. Analysis of the USH2A transcripts of these cells revealed that one of the patient's mutations causes exonification of intron 40, a translation frameshift and a premature stop codon. Western blotting revealed upregulation of GRP78 and GRP94, suggesting that the patient's other USH2A variant (Arg4192His) causes disease through protein misfolding and ER stress. Transplantation into 4-day-old immunodeficient Crb1 (-/-) mice resulted in the formation of morphologically and immunohistochemically recognizable photoreceptor cells, suggesting that the mutations in this patient act via post-developmental photoreceptor degeneration. DOI:http://dx.doi.org/10.7554/eLife.00824.001. | | 23991284
|
Efficient integration of transgenes into a defined locus in human embryonic stem cells.
Kenji Sakurai,Miho Shimoji,Candice G T Tahimic,Kazuhiro Aiba,Eihachiro Kawase,Kouichi Hasegawa,Yuji Amagai,Hirofumi Suemori,Norio Nakatsuji
Nucleic acids research
38
2010
Show Abstract
Random integration is one of the more straightforward methods to introduce a transgene into human embryonic stem (ES) cells. However, random integration may result in transgene silencing and altered cell phenotype due to insertional mutagenesis in undefined gene regions. Moreover, reliability of data may be compromised by differences in transgene integration sites when comparing multiple transgenic cell lines. To address these issues, we developed a genetic manipulation strategy based on homologous recombination and Cre recombinase-mediated site-specific integration. First, we performed gene targeting of the hypoxanthine phosphoribosyltransferase 1 (HPRT) locus of the human ES cell line KhES-1. Next, a gene-replacement system was created so that a circular vector specifically integrates into the targeted HPRT locus via Cre recombinase activity. We demonstrate the application of this strategy through the creation of a tetracycline-inducible reporter system at the HPRT locus. We show that reporter gene expression was responsive to doxycycline and that the resulting transgenic human ES cells retain their self-renewal capacity and pluripotency. Full Text Article | | 20071742
|
Role of Pax6 in development of the cerebellar system.
Engelkamp, D, et al.
Development, 126: 3585-96 (1999)
1999
Show Abstract
Post-mitotic neurons generated at the rhombic lip undertake long distance migration to widely dispersed destinations, giving rise to cerebellar granule cells and the precerebellar nuclei. Here we show that Pax6, a key regulator in CNS and eye development, is strongly expressed in rhombic lip and in cells migrating away from it. Development of some structures derived from these cells is severely affected in Pax6-null Small eye (Pax6(Sey)/Pax6(Sey)) embryos. Cell proliferation and initial differentiation seem unaffected, but cell migration and neurite extension are disrupted in mutant embryos. Three of the five precerebellar nuclei fail to form correctly. In the cerebellum the pre-migratory granule cell sub-layer and fissures are absent. Some granule cells are found in ectopic positions in the inferior colliculus which may result from the complete absence of Unc5h3 expression in Pax6(Sey)/Pax6(Sey) granule cells. Our results suggest that Pax6 plays a strong role during hindbrain migration processes and at least part of its activity is mediated through regulation of the netrin receptor Unc5h3. | | 10409504
|