Role of anoctamin-1 and bestrophin-1 in spinal nerve ligation-induced neuropathic pain in rats.
Pineda-Farias, JB; Barragán-Iglesias, P; Loeza-Alcocer, E; Torres-López, JE; Rocha-González, HI; Pérez-Severiano, F; Delgado-Lezama, R; Granados-Soto, V
Molecular pain
11
41
2015
Show Abstract
Calcium-activated chloride channels (CaCCs) activation induces membrane depolarization by increasing chloride efflux in primary sensory neurons that can facilitate action potential generation. Previous studies suggest that CaCCs family members bestrophin-1 and anoctamin-1 are involved in inflammatory pain. However, their role in neuropathic pain is unclear. In this investigation we assessed the involvement of these CaCCs family members in rats subjected to the L5/L6 spinal nerve ligation. In addition, anoctamin-1 and bestrophin-1 mRNA and protein expression in dorsal root ganglion (DRG) and spinal cord was also determined in the presence and absence of selective inhibitors.L5/L6 spinal nerve ligation induced mechanical tactile allodynia. Intrathecal administration of non-selective CaCCs inhibitors (NPPB, 9-AC and NFA) dose-dependently reduced tactile allodynia. Intrathecal administration of selective CaCCs inhibitors (T16Ainh-A01 and CaCCinh-A01) also dose-dependently diminished tactile allodynia and thermal hyperalgesia. Anoctamin-1 and bestrophin-1 mRNA and protein were expressed in the dorsal spinal cord and DRG of naïve, sham and neuropathic rats. L5/L6 spinal nerve ligation rose mRNA and protein expression of anoctamin-1, but not bestrophin-1, in the dorsal spinal cord and DRG from day 1 to day 14 after nerve ligation. In addition, repeated administration of CaCCs inhibitors (T16Ainh-A01, CaCCinh-A01 or NFA) or anti-anoctamin-1 antibody prevented spinal nerve ligation-induced rises in anoctamin-1 mRNA and protein expression. Following spinal nerve ligation, the compound action potential generation of putative C fibers increased while selective CaCCs inhibitors (T16Ainh-A01 and CaCCinh-A01) attenuated such increase.There is functional anoctamin-1 and bestrophin-1 expression in rats at sites related to nociceptive processing. Blockade of these CaCCs suppresses compound action potential generation in putative C fibers and lessens established tactile allodynia. As CaCCs activity contributes to neuropathic pain maintenance, selective inhibition of their activity may function as a tool to generate analgesia in nerve injury pain states. | | | 26130088
 |
AMPA Receptor-mTOR Activation is Required for the Antidepressant-Like Effects of Sarcosine during the Forced Swim Test in Rats: Insertion of AMPA Receptor may Play a Role.
Chen, KT; Tsai, MH; Wu, CH; Jou, MJ; Wei, IH; Huang, CC
Frontiers in behavioral neuroscience
9
162
2015
Show Abstract
Sarcosine, an endogenous amino acid, is a competitive inhibitor of the type I glycine transporter and an N-methyl-d-aspartate receptor (NMDAR) coagonist. Recently, we found that sarcosine, an NMDAR enhancer, can improve depression-related behaviors in rodents and humans. This result differs from previous studies, which have reported antidepressant effects of NMDAR antagonists. The mechanisms underlying the therapeutic response of sarcosine remain unknown. This study examines the role of mammalian target of rapamycin (mTOR) signaling and α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor (AMPAR) activation, which are involved in the antidepressant-like effects of several glutamatergic system modulators. The effects of sarcosine in a forced swim test (FST) and the expression levels of phosphorylated mTOR signaling proteins were examined in the absence or presence of mTOR and AMPAR inhibitors. In addition, the influence of sarcosine on AMPAR trafficking was determined by analyzing the phosphorylation of AMPAR subunit GluR1 at the PKA site (often considered an indicator for GluR1 membrane insertion in neurons). A single injection of sarcosine exhibited antidepressant-like effects in rats in the FST and rapidly activated the mTOR signaling pathway, which were significantly blocked by mTOR inhibitor rapamycin or the AMPAR inhibitor 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline (NBQX) pretreatment. Moreover, NBQX pretreatment eliminated the ability of sarcosine to stimulate the phosphorylated mTOR signaling proteins. Furthermore, GluR1 phosphorylation at its PKA site was significantly increased after an acute in vivo sarcosine treatment. The results demonstrated that sarcosine exerts antidepressant-like effects by enhancing AMPAR-mTOR signaling pathway activity and facilitating AMPAR membrane insertion. Highlights-A single injection of sarcosine rapidly exerted antidepressant-like effects with a concomitant increase in the activation of the mammalian target of rapamycin mTOR signaling pathway.-The antidepressant-like effects of sarcosine occur through the activated AMPAR-mTOR signaling pathway.-Sarcosine could enhance AMPAR membrane insertion via an AMPAR throughput. | | | 26150775
|
Autophagy Regulates Formation of Primary Cilia in Mefloquine-Treated Cells.
Shin, JH; Bae, DJ; Kim, ES; Kim, HB; Park, SJ; Jo, YK; Jo, DS; Jo, DG; Kim, SY; Cho, DH
Biomolecules & therapeutics
23
327-32
2015
Show Abstract
Primary cilia have critical roles in coordinating multiple cellular signaling pathways. Dysregulation of primary cilia is implicated in various ciliopathies. To identify specific regulators of autophagy, we screened chemical libraries and identified mefloquine, an anti-malaria medicine, as a potent regulator of primary cilia in human retinal pigmented epithelial (RPE) cells. Not only ciliated cells but also primary cilium length was increased in mefloquine-treated RPE cells. Treatment with mefloquine strongly induced the elongation of primary cilia by blocking disassembly of primary cilium. In addition, we found that autophagy was increased in mefloquine-treated cells by enhancing autophagic flux. Both chemical and genetic inhibition of autophagy suppressed ciliogenesis in mefloquine-treated RPE cells. Taken together, these results suggest that autophagy induced by mefloquine positively regulates the elongation of primary cilia in RPE cells. | | | 26157548
|
VPS35 pathogenic mutations confer no dominant toxicity but partial loss of function in Drosophila and genetically interact with parkin.
Malik, BR; Godena, VK; Whitworth, AJ
Human molecular genetics
24
6106-17
2015
Show Abstract
Mutations in VPS35 (PARK17) cause autosomal dominant, late onset Parkinson's disease (PD). VPS35 forms a core component of the retromer complex that mediates the retrieval of membrane proteins from endosomes back to either the Golgi or plasma membrane. While aberrant endosomal protein sorting has been linked to several neurodegenerative diseases, the mechanisms by which VPS35 mutations and retromer function contribute to PD pathogenesis are not clear. To address this, we generated transgenic Drosophila that express variant forms of human VPS35 found in PD cases and the corresponding variants of the Drosophila ortholog. We did not find evidence of dominant toxicity from any variant form including the pathogenic D620N mutation, even with aging. However, assessing the ability of Vps35 variants to rescue multiple vps35-mutant phenotypes, we found that the D620N mutation confers a partial loss of function. Recently, VPS35 has been linked to the formation of mitochondria-derived vesicles, which mediate the degradation of mitochondrial proteins and contribute to mitochondrial quality control. This process is also promoted by two other PD-lined genes parkin (PARK2) and PINK1 (PARK6). We demonstrate here that vps35 genetically interacts with parkin but interestingly not with pink1. Strikingly, Vps35 overexpression is able to rescue several parkin-mutant phenotypes. Together these findings provide in vivo evidence that the D620N mutation likely confers pathogenicity through a partial loss of function mechanism and that this may be linked to other known pathogenic mechanisms such as mitochondrial dysfunction. | | | 26251041
|
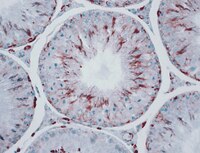
Proteins from formalin-fixed paraffin-embedded prostate cancer sections that predict the risk of metastatic disease.
Dunne, JC; Lamb, DS; Delahunt, B; Murray, J; Bethwaite, P; Ferguson, P; Nacey, JN; Sondhauss, S; Jordan, TW
Clinical proteomics
12
24
2015
Show Abstract
Prostate cancer is the most frequently diagnosed cancer in men and the third leading cause of cancer related deaths among men living in developed countries. Biomarkers that predict disease outcome at the time of initial diagnosis would substantially aid disease management.Proteins extracted from formalin-fixed paraffin-embedded tissue were identified using nanoflow liquid chromatography-MALDI MS/MS or after separation by one- or two-dimensional electrophoresis. The proteomics data have been deposited to the ProteomeXchange with identifier PXD000963. A list of potential biomarker candidates, based on proposed associations with prostate cancer, was derived from the 320 identified proteins. Candidate biomarkers were then examined by multiplexed Western blotting of archival specimens from men with premetastatic disease and subsequent disease outcome data. Annexin A2 provided the best prediction of risk of metastatic disease (log-rank Chi squared p = 0. 025). A tumor/control tissue greater than 2-fold relative abundance increase predicted early biochemical failure, while less than 2-fold change predicted late or no biochemical failure.This study confirms the potential for use of archival FFPE specimens in the search for prognostic biomarkers for prostate cancer and suggests that annexin A2 abundance in diagnostic biopsies is predictive for metastatic potential. Protein profiling each cancer may lead to an overall reduction in mortality from metastatic prostate cancer as well as reduced treatment associated morbidity. | | | 26388710
|
Insulin demand regulates β cell number via the unfolded protein response.
Sharma, RB; O'Donnell, AC; Stamateris, RE; Ha, B; McCloskey, KM; Reynolds, PR; Arvan, P; Alonso, LC
The Journal of clinical investigation
125
3831-46
2015
Show Abstract
Although stem cell populations mediate regeneration of rapid turnover tissues, such as skin, blood, and gut, a stem cell reservoir has not been identified for some slower turnover tissues, such as the pancreatic islet. Despite lacking identifiable stem cells, murine pancreatic β cell number expands in response to an increase in insulin demand. Lineage tracing shows that new β cells are generated from proliferation of mature, differentiated β cells; however, the mechanism by which these mature cells sense systemic insulin demand and initiate a proliferative response remains unknown. Here, we identified the β cell unfolded protein response (UPR), which senses insulin production, as a regulator of β cell proliferation. Using genetic and physiologic models, we determined that among the population of β cells, those with an active UPR are more likely to proliferate. Moreover, subthreshold endoplasmic reticulum stress (ER stress) drove insulin demand-induced β cell proliferation, through activation of ATF6. We also confirmed that the UPR regulates proliferation of human β cells, suggesting that therapeutic UPR modulation has potential to expand β cell mass in people at risk for diabetes. Together, this work defines a stem cell-independent model of tissue homeostasis, in which differentiated secretory cells use the UPR sensor to adapt organ size to meet demand. | | | 26389675
|
Inhibition of autophagy suppresses sertraline-mediated primary ciliogenesis in retinal pigment epithelium cells.
Kim, ES; Shin, JH; Park, SJ; Jo, YK; Kim, JS; Kang, IH; Nam, JB; Chung, DY; Cho, Y; Lee, EH; Chang, JW; Cho, DH
PloS one
10
e0118190
2015
Show Abstract
Primary cilia are conserved cellular organelles that regulate diverse signaling pathways. Autophagy is a complex process of cellular degradation and recycling of cytoplasmic proteins and organelles, and plays an important role in cellular homeostasis. Despite its potential importance, the role of autophagy in ciliogenesis is largely unknown. In this study, we identified sertraline as a regulator of autophagy and ciliogenesis. Sertraline, a known antidepressant, induced the growth of cilia and blocked the disassembly of cilia in htRPE cells. Following treatment of sertraline, there was an increase in the number of cells with autophagic puncta and LC3 protein conversion. In addition, both a decrease of ATG5 expression and the treatment of an autophagy inhibitor resulted in the suppression of the sertraline-induced activation of autophagy in htRPE cells. Interestingly, we found that genetic and chemical inhibition of autophagy attenuated the growth of primary cilia in htRPE cells. Taken together, our results suggest that the inhibition of autophagy suppresses sertraline-induced ciliogenesis. | | | 25671433
|
Novel Mechanisms of Spinal Cord Plasticity in a Mouse Model of Motoneuron Disease.
Gulino, R; Parenti, R; Gulisano, M
BioMed research international
2015
654637
2015
Show Abstract
A hopeful spinal cord repairing strategy involves the activation of neural precursor cells. Unfortunately, their ability to generate neurons after injury appears limited. Another process promoting functional recovery is synaptic plasticity. We have previously studied some mechanisms of spinal plasticity involving BDNF, Shh, Notch-1, Numb, and Noggin, by using a mouse model of motoneuron depletion induced by cholera toxin-B saporin. TDP-43 is a nuclear RNA/DNA binding protein involved in amyotrophic lateral sclerosis. Interestingly, TDP-43 could be localized at the synapse and affect synaptic strength. Here, we would like to deepen the investigation of this model of spinal plasticity. After lesion, we observed a glial reaction and an activity-dependent modification of Shh, Noggin, and Numb proteins. By using multivariate regression models, we found that Shh and Noggin could affect motor performance and that these proteins could be associated with both TDP-43 and Numb. Our data suggest that TDP-43 is likely an important regulator of synaptic plasticity, probably in collaboration with other proteins involved in both neurogenesis and synaptic plasticity. Moreover, given the rapidly increasing knowledge about spinal cord plasticity, we believe that further efforts to achieve spinal cord repair by stimulating the intrinsic potential of spinal cord will produce interesting results. | | | 26064939
|
Recognition of Linear B-Cell Epitope of Betanodavirus Coat Protein by RG-M18 Neutralizing mAB Inhibits Giant Grouper Nervous Necrosis Virus (GGNNV) Infection.
Chen, CW; Wu, MS; Huang, YJ; Cheng, CA; Chang, CY
PloS one
10
e0126121
2015
Show Abstract
Betanodavirus is a causative agent of viral nervous necrosis syndrome in many important aquaculture marine fish larvae, resulting in high global mortality. The coat protein of Betanodavirus is the sole structural protein, and it can assemble the virion particle by itself. In this study, we used a high-titer neutralizing mAB, RG-M18, to identify the linear B-cell epitope on the viral coat protein. By mapping a series of recombinant proteins generated using the E. coli PET expression system, we demonstrated that the linear epitope recognized by RG-M18 is located at the C-terminus of the coat protein, between amino acid residues 195 and 338. To define the minimal epitope region, a set of overlapping peptides were synthesized and evaluated for RG-M18 binding. Such analysis identified the 195VNVSVLCR202 motif as the minimal epitope. Comparative analysis of Alanine scanning mutagenesis with dot-blotting and ELISA revealed that Valine197, Valine199, and Cysteine201 are critical for antibody binding. Substitution of Leucine200 in the RGNNV, BFNNV, and TPNNV genotypes with Methionine200 (thereby simulating the SJNNV genotype) did not affect binding affinity, implying that RG-M18 can recognize all genotypes of Betanodaviruses. In competition experiments, synthetic multiple antigen peptides of this epitope dramatically suppressed giant grouper nervous necrosis virus (GGNNV) propagation in grouper brain cells. The data provide new insights into the protective mechanism of this neutralizing mAB, with broader implications for Betanodavirus vaccinology and antiviral peptide drug development. | | | 25938761
|
Prdx4 is a compartment-specific H2O2 sensor that regulates neurogenesis by controlling surface expression of GDE2.
Yan, Y; Wladyka, C; Fujii, J; Sockanathan, S
Nature communications
6
7006
2015
Show Abstract
Neural progenitors and terminally differentiated neurons show distinct redox profiles, suggesting that coupled-redox cascades regulate the initiation and progression of neuronal differentiation. Discrete cellular compartments have different redox environments and how they contribute to differentiation is unclear. Here we show that Prdx4, an endoplasmic reticulum (ER) enzyme that metabolizes H2O2, acts as a tunable regulator of neurogenesis via its compartmentalized thiol-oxidative function. Prdx4 ablation causes premature motor neuron differentiation and progenitor depletion, leading to imbalances in subtype-specific motor neurons. GDE2, a six-transmembrane protein that induces differentiation by downregulating Notch signalling through surface cleavage of GPI-anchored proteins, is targeted by Prdx4 oxidative activity. Prdx4 dimers generated by H2O2 metabolism oxidize two cysteine residues within the GDE2 enzymatic domain, which blocks GDE2 trafficking to the plasma membrane and prevents GDE2 neurogeneic function. Thus, Prdx4 oxidative activity acts as a sensor to directly couple neuronal differentiation with redox environments in the ER. | | | 25943695
|